Einführung
Laut FDA „bezieht sich der Begriff Qualifizierung auf Aktivitäten, die durchgeführt werden, um nachzuweisen, dass Betriebsmittel und Ausrüstung für die beabsichtigte Verwendung geeignet sind und ordnungsgemäß funktionieren. Diese Aktivitäten gehen notwendigerweise der Herstellung von Produkten im kommerziellen Maßstab voraus“.
Die Qualifizierung kann in drei Phasen unterteilt werden: Installationsqualifizierung (IQ), Betriebsqualifizierung (OQ) und Leistungsqualifizierung (PQ).IQ, OQ, PQ ist für die Gerätequalifizierung unerlässlich.
Die Validierung und die damit verbundenen Qualifizierungsschritte sind in verschiedenen Teilen von 21 CFR 211 (Arzneimittel) und 21 CFR 820 (Medizinprodukte) geregelt und werden von der FDA durchgesetzt
Diese Geräte befinden sich an verschiedenen Stellen der Lieferkette, von komplexen, futuristischen Labors bis hin zu Amazon-ähnlichen Verpackungsanlagen. Eines haben sie jedoch gemeinsam: Ein Ausfall eines dieser Geräte an einem beliebigen Punkt der Kette kann zu einem gefährlichen Produktfehler führen – falsch etikettierte Produkte, falsche Dosierung, falsche Formulierung, nicht versiegelte oder unhygienische Produkte und mehr.
Die Botschaft ist klar: Pharmaunternehmen müssen der Qualitätssicherung bei der Installation und Verwendung von Geräten die gleiche Aufmerksamkeit schenken wie bei der Formulierung und dem Vertrieb ihrer lebensrettenden Produkte – Arzneimittel, Impfstoffe und therapeutische Produkte.
Der Goldstandard der Qualitätssicherung von Anlagen wird mit IQ OQ PQ abgekürzt. Dieser Prozess wendet Qualitätssicherungsstandards auf kritische Geräte in drei Phasen des Installationsprozesses an. IQ OQ PQ ist eine wichtige Hürde, die ein Pharmaunternehmen nehmen muss, um die gesetzlichen Vorschriften zu erfüllen.
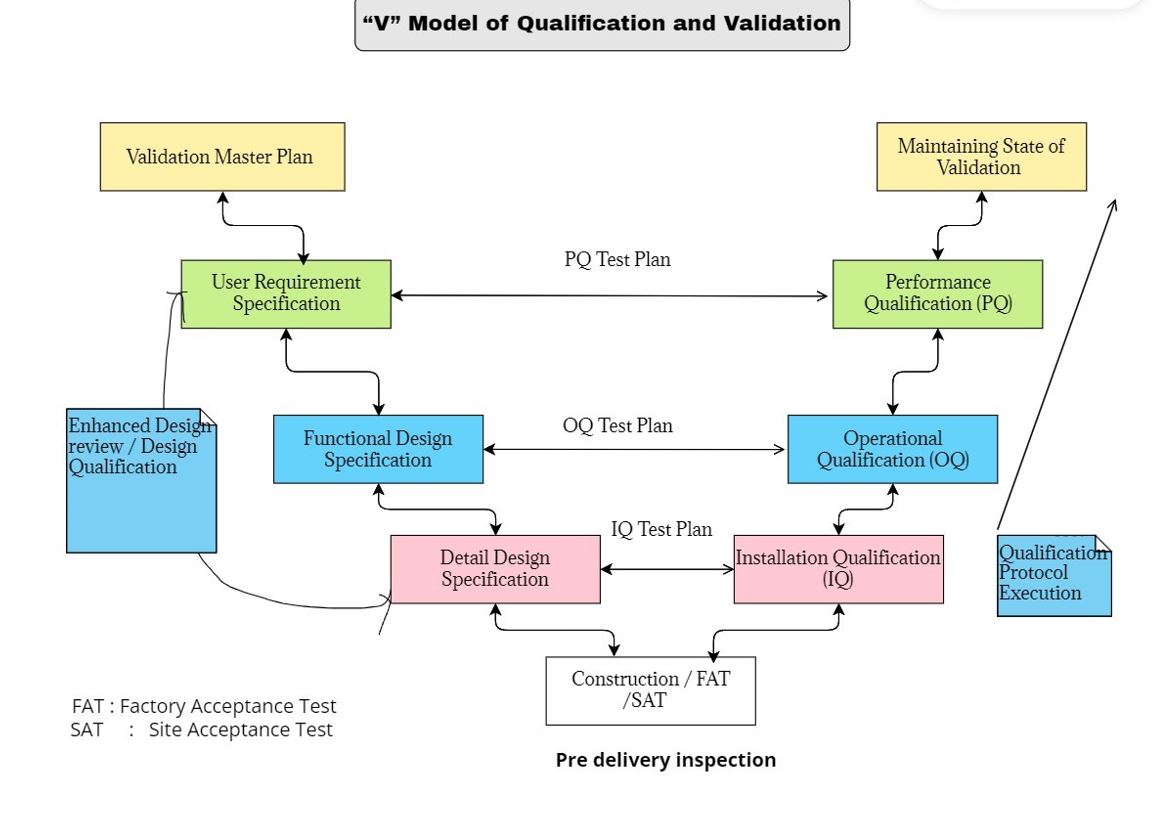
Was ist das V-Modell ?
Das V-Modell ist eine Methode zur Visualisierung und zum Vergleich der Beziehung zwischen den Benutzeranforderungen, dem funktionalen Entwurf und dem Feinentwurf und der darauf basierenden Installationsqualifizierung (IQ), Betriebsqualifizierung (OQ) und Leistungsqualifizierung (PQ) (siehe Abbildung unten).
Was ist IQ OQ PQ in der pharmazeutischen Fertigungsindustrie?
IQ OQ PQ steht für Installation Qualification, Operational Qualification und Performance Qualification. Dabei handelt es sich um drei aufeinander aufbauende Prozesse, die dazu dienen, zu bestätigen und zu dokumentieren, dass die Ausrüstung, die Rohrleitungen, die Instrumente und die Versorgungseinrichtungen (Luft, Wasser, Dampf) usw. korrekt installiert sind, wie beabsichtigt funktionieren und durchgängig gemäß den Konstruktionsspezifikationen funktionieren.
Das Ziel dieser Prozesse ist die Erstellung eines dokumentierten Nachweises (auf Papier oder elektronisch) mit mehreren Unterschriften aus allen relevanten Abteilungen (Validierung, Technik, Wartung, Kalibrierung, Qualitätssicherung usw.). Diese Dokumentation beweist den (Eigentümern/Auftraggebern oder Aufsichtsbehörden), dass die bestellte kritische Ausrüstung geliefert, installiert und korrekt konfiguriert wurde und dass das System als Ganzes gemäß den technischen Zeichnungen und Konstruktionsspezifikationen funktioniert.
Das GMP-System erfordert die Praktiken der IQ, OQ und PQ für den Qualifizierungsprozess der Ausrüstung. Es hilft den Herstellern, eine gleichbleibende Qualität der Ausrüstung zu gewährleisten. Außerdem werden durch all diese Verfahren Fehler erheblich reduziert, so dass die Produktqualität in Übereinstimmung mit den einschlägigen Vorschriften und Industrienormen aufrechterhalten werden kann. Sie können die Gerätequalifizierung intern durchführen oder sich an Fachleute wenden, die Dienstleistungen zur Validierung von Geräten anbieten.
Sie benötigen fachkundige Beratung bei Projekte zur Gerätequalifizierung ?
Warum brauchen wir Qualifizierung (IQ OQ PQ ) in der lebenswissenschaftlichen Industrie?
Die Qualifizierung wird in der Regel von der technischen Gruppe, dem Validierungsteam oder einer anderen Person oder Gruppe durchgeführt, die über die erforderlichen Qualifikationen und Kenntnisse in Bezug auf die Verwendung und den Betrieb des Geräts verfügt und die für die Durchführung der erforderlichen Aufgaben geschult und erfahren ist.
Dafür gibt es zwei Hauptgründe.
1) Der erste Grund ist, dass es eine gesetzliche Vorschrift ist.
Nach Angaben der Food and Drug Administration (FDA)
„Geräte, die bei der Herstellung, Verarbeitung, Verpackung oder Aufbewahrung eines Arzneimittels verwendet werden, müssen von geeigneter Konstruktion, angemessener Größe und an geeigneter Stelle angebracht sein, um den Betrieb für den vorgesehenen Verwendungszweck sowie die Reinigung und Wartung zu erleichtern.“ – 21 CFR Titel 211.63,
Innerhalb der EU enthält EudraLex – Band 4 – Gute Herstellungspraxis (GMP) unter Anhang 15: Qualifizierung und Validierung eine ausführlichere Anleitung zur Qualifizierung.
Und obwohl es sich nicht um eine Vorschrift handelt, ist der ISPE Baseline Guide 5 Commissioning and Qualification (Second Edition) ein weit verbreiteter Leitfaden für die Qualifizierung, der von zahlreichen Regulierungsbehörden unterstützt wird.
2) Der zweite Grund ist, dass sich bei der Qualifizierung und Validierung einer neuen Anlage oder eines neuen Verfahrens selbst der kleinste Installationsfehler oder das trivialste Problem mit der Leistung der Ausrüstung zu einem ernsthaften Produktqualitätsproblem mit tödlichen Folgen für die Patienten auswachsen kann. Dieses Problem ist besonders akut bei neuartigen oder neuen Systemen, bei denen es keinerlei Erfahrungen mit der Leistung oder dem Ausfall gibt und selbst kleinste Probleme zu kranken oder toten Patienten führen können.
Was ist die Installationsqualifizierung (IQ)?
Die Installationsqualifizierung (IQ) ist ein kritischer, dokumentierter Prozess, mit dem sichergestellt wird, dass Geräte, Rohrleitungen, Software oder Instrumente, die für die Produktqualität wichtig sind, korrekt geliefert, installiert und konfiguriert werden. Dieser Prozess dient als gründliche Überprüfung anhand einer vorgegebenen Checkliste, um sicherzustellen, dass alle installierten Komponenten mit den Konstruktionsspezifikationen und Installationsstandards übereinstimmen, unabhängig davon, ob diese vom Hersteller oder durch eine genehmigte Checkliste festgelegt wurden.
Stellen Sie sich vor, Sie installieren eine neue Maschine; IQ hilft Ihnen zu bestätigen, dass nicht nur die richtigen Geräte geliefert und aus den vorgeschriebenen Materialien hergestellt wurden, sondern dass sie auch an der richtigen Stelle installiert sind und alle Anschlüsse, wie Rohrleitungen und elektrische Leitungen, ordnungsgemäß eingerichtet sind, und dass die von Ihnen in den detaillierten Konstruktionsspezifikationen angegebenen Instrumente korrekt geliefert und installiert wurden. Diese detaillierte Prüfung wird in der Regel von Fachleuten wie Inbetriebnahme-, Qualifizierungs- und Validierungsingenieuren (CQV), Inbetriebnahme- und Qualifizierungsingenieuren (C&Q) oder Validierungstechnikern durchgeführt.
Dies ist der erste Schritt. Das IQ-Dokument für die Ausrüstung wird von der technischen Abteilung in Abstimmung mit dem Benutzer und der QA erstellt. Die IQ wird durchgeführt, um sicherzustellen, dass die Räumlichkeiten, die die Versorgungseinrichtungen und Geräte unterstützen, in Übereinstimmung mit den genehmigten Konstruktionsspezifikationen (DQ) und den Herstellerhandbüchern und -empfehlungen gebaut und installiert wurden.
Was macht IQ erfolgreich?
Vergleicht das, was „angegeben“ wurde, mit dem, was vor Ort „vorgefunden“ wurde. Alle bei der Installationsqualifizierung festgestellten Abweichungen sind zu protokollieren.
- Abgleich der Lieferung mit den Packlisten und Überprüfung auf Schäden.
- Sicherstellung, dass die Ausrüstungsspezifikationen mit den Entwurfs- und Betriebsanforderungen übereinstimmen.
- Überprüfung der Richtigkeit der Seriennummern, der Marke und des Modells sowie der korrekten Identifizierung der Komponenten.
- Überprüfung der korrekten Installationsorte, der Anschlüsse an andere Geräte und der Installation von Sekundär- und Zusatzgeräten.
- Überprüfen der Energie- oder Hilfsmittelversorgung und Sicherstellen, dass die Umgebungs- und Betriebsbedingungen den Herstellerrichtlinien entsprechen.
- Überprüfung von Software-Installationen und Aufzeichnung von Firmware-Versionen und Seriennummern.
- Dokumentation der Kalibrierungs- und Validierungsdaten und Sammlung aller zugehörigen Handbücher und Zertifikate.

Was ist Operative Qualifizierung (OQ)?
Die Betriebsqualifizierung (OQ) stellt sicher, dass die Ausrüstung den Anforderungen des Betreibers entspricht und innerhalb der vom Hersteller definierten Bereiche arbeitet. Die OQ, die für die Abnahme von technischen Geräten und Anlagen unerlässlich ist, schreibt die Prüfung einzelner Komponenten gemäß einem Prüfplan mit detaillierter Leistungsdokumentation vor.
In dieser Phase, die nach der IQ beginnt, werden die Betriebssicherheit der Ausrüstung und die Einhaltung der Betriebsspezifikationen bestätigt. Die Requalifizierung, die nach Ausrüstungsänderungen, größeren Wartungsarbeiten oder als Teil der Qualitätssicherung von entscheidender Bedeutung ist, konzentriert sich auf die Bewertung von Ausrüstungsattributen, die für die Produktqualität entscheidend sind, und stärkt die Rolle der OQ bei der Validierung des Beitrags der Ausrüstung zur Integrität des Endprodukts.
Was macht Operation Qualification (OQ) erfolgreich?
Hier wird das, was im Funktionsentwurf „festgelegt“ wurde, mit den Testergebnissen verglichen.
- Nach erfolgreichem Abschluss der IQ. Die OQ wird durchgeführt, um zu verifizieren, dass die Ausrüstung, das Instrument, das Hilfsmittel und das System in Bezug auf die Konstruktionsspezifikation und die cGMP-Anforderungen funktionieren.
- Es ist zu prüfen und zu dokumentieren, dass die Geräte und Systeme wie vorgesehen funktionieren und innerhalb der vom Hersteller angegebenen Betriebsbereiche liegen.
- Betriebstests sind wann immer möglich durchzuführen, um das System bis an die Grenzen der erwarteten Betriebsbedingungen, der Alarmverriegelung und der grundlegenden Funktionen gemäß der Funktionsspezifikation der Maschine zu testen.
- Normalerweise wird die OQ ohne Last durchgeführt. Kann die Anlage jedoch nicht ohne Last betrieben werden, so können Lastversuche durchgeführt werden.
- Bei der OQ muss auch die Leistung der Ausrüstungskomponenten überprüft werden, wie z. B. Motoren, Gebläse, Sensoren, funktionierende Verriegelungen, Sicherheitseinrichtungen usw.
- Jede während der Betriebsqualifikation festgestellte Nichtübereinstimmung ist zu protokollieren.
- Nach erfolgreichem Abschluss der OQ sollten die Betriebs- und Reinigungsverfahren, die Bedienerschulung und die Anforderungen an die vorbeugende Wartung festgelegt werden.
- Im Protokoll nach dem Prüfplan sind Verweise auf eventuelle Abweichungen von den vordefinierten Spezifikationen und auf eventuelle Änderungskontrollen hinzuzufügen.
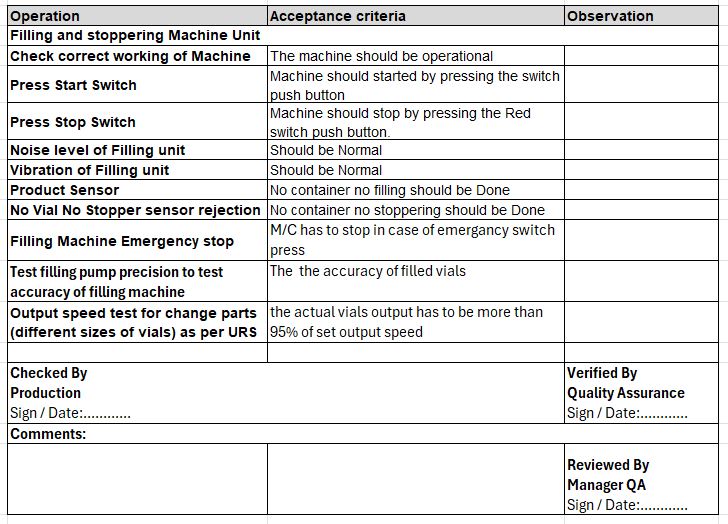
Beispiel: Betrachten Sie eine Fläschchenabfüllmaschine, lassen Sie uns die grundlegende Betriebsqualifikationsprüfung und die Abnahmekriterien überprüfen.
Was ist Leistungsqualifizierung (PQ)?
Die Leistungsqualifizierung bestätigt, dass die Geräte und Systeme den Anforderungen der Benutzer entsprechen und für die beabsichtigte Verwendung geeignet sind, wie sie in der Benutzerspezifikation (URS) definiert ist.
Die PQ sollte normalerweise auf den erfolgreichen Abschluss von IQ und OQ folgen. Durch die PQ können Organisationen mit konkreten Nachweisen belegen, dass ihre Ausrüstung nicht nur die Konstruktionsspezifikationen und gesetzlichen Anforderungen erfüllt, sondern auch unter tatsächlichen Betriebsbedingungen durchgängig hochwertige Produkte erzeugt.
Was macht Performance Qualification (PQ) erfolgreich?

Beachten Sie bei der Durchführung der PQ die folgenden Punkte,
- Die PQ verifiziert die URS (Benutzeranforderungsspezifikation) mit dem PQ-Prüfplan und bestätigt und dokumentiert, dass die Geräte und Systeme für die vorgesehene Verwendung geeignet sind. Dies ist der letzte Schritt der Gerätequalifizierung.
- Die Tests sollten den Betriebsbereich des beabsichtigten Prozesses abdecken, wobei die Ausrüstung unter Last steht.
- Sie ähnelt der Betriebsqualifizierung, da sie die Betriebsanforderungen der Ausrüstung testet, aber in diesem Fall enthält die Ausrüstung eine Last oder ein Prozessfüllmedium, um das reale Produktionsszenario zu testen.
- Mit anderen Worten: Die Anlage wird unter realen Bedingungen getestet, d. h. unter den Bedingungen, denen die Anlage während der Serienproduktion ausgesetzt ist.
- Diese Phase ist von großer Bedeutung, da sie die Funktionsweise, die Kräfte und die Energie der einzelnen Komponenten der Anlage zu einem harmonischen System vereint. Dabei können in dieser Phase der Qualifizierung Fehler festgestellt werden wie
- Normalerweise sind Präqualifikationsanforderungen wie SOP für den Betrieb, SOP für die vorbeugende Instandhaltung, Schulungsunterlagen der Validierungsteams.
- In der PQ wird nachgewiesen, dass das System wie vorgesehen funktioniert, indem es wiederholt nach den vorgesehenen Plänen unter normalen Betriebsbedingungen und unter Worst-Case-Bedingungen betrieben wird.
Zum Beispiel: Für eine Abfüll- und Verschließmaschine. Die PQ helfen dabei, die Fähigkeit der Maschine zur Abfüllung gleichmäßiger Volumina bei verschiedenen Laufgeschwindigkeiten und zur Überprüfung des Verschließens von Fläschchen anhand des unten stehenden Testplans in Bezug auf URS zu ermitteln.
Schlussfolgerung:
- IQ-, OQ- und PQ-Aktivitäten sind obligatorisch, um die GMP-Vorschriften einzuhalten, ebenso wie Inbetriebnahmeaktivitäten. Diese robuste Inbetriebnahmestrategie stellt sicher, dass Ihre Anlagen, Geräte und Versorgungseinrichtungen ordnungsgemäß installiert wurden und gemäß den vorgegebenen Bedingungen funktionieren.
- Eine GMP-Auswirkungsanalyse ist ein entscheidender Schritt für die CQV-Aktivitäten, da sie zwischen direkten, indirekten und nicht-auswirkenden Systemen sowie kritischen und nicht-kritischen Komponenten unterscheidet und so den Aufwand für die Inbetriebnahme und die Qualifizierung und Validierung optimiert.
- CQV-Aktivitäten sind keine einmalige Angelegenheit. Es müssen regelmäßige Überprüfungs-/Requalifizierungsroutinen eingeführt werden, um den Systemstatus, die zugehörigen Änderungskontrollen und Abweichungen oder die Ergebnisse der Betriebskontrollen zu überprüfen, um Trends zu erkennen und Präventivmaßnahmen zur Vermeidung künftiger Abweichungen zu ergreifen.
Brauchen Sie Hilfe?
Benötigen Sie fachkundige Beratung zur Qualifizierung und Validierung?
Ist Ihr Team auf der Suche nach spezialisierten, motivierten Mitarbeitern für eine effektive Validierung? Wir unterstützen Sie bei der Einführung robuster, rechtskonformer Systeme, die auf Ihre spezifischen Bedürfnisse zugeschnitten sind.
Wenden Sie sich noch heute an uns, um Ihren Prozess gemeinsam abzustimmen. Mit der richtigen Strategie wird die Validierung von Computersystemen für Sie zur Nebensache.
Nützlicher Link:
- EudraLex, Volume 4, EU Guidelines for Good Manufacturing Practice, Annex 15: Qualification and Validation
- 21 CFR Part 210 – Current Good Manufacturing Practice in Manufacturing. Processing, Packing, or Holding of Drug
- 21 CFR Part 211 – Current Good Manufacturing Practice for Finished Pharmaceuticals 4. TGA – PE009, the PIC/S guide to GMP for medicinal products 5. ICH Q9: Quality Risk Management, Step 5 version
- ISPE Baseline Guide Volume 5: Commissioning and Qualification
- ISPE GAMP 5: A Risk-Based Approach to Compliant GxP Computerized Systems
- https://www.linkedin.com/in/marco-klinger-b86bb3151/recent-activity/all/

Sagar Pawar
Computer System Validation Specialist


