
USFDA warning letters explained in this blog. Every year, the FDA issues hundreds of warning letters along with inspections of manufacturing facilities. You should take the warning letters seriously because they act as a precursor to a worse failure. You should always take proper corrective and preventative actions accordingly and make a comprehensive gap analysis based on the mentioned requirements to be fully compliant.
FDA warning letters typically follow inspections where deviations or violations of regulations, such as those related to cGMP, data integrity etc. During inspections, auditors document these deviations using Form 483. While Forms 483 and FDA Warning Letters are distinct, the warning letter often follows as the next step in the regulatory process..
The FDA warning letter describes the violation and the potential problem, followed by specifying what evidence the manufacturer must present to document the addressed concern.
When a manufacturer significantly violates FDA regulations, they receive a Warning Letter from the FDA outlining the violation, such as poor manufacturing practices or inaccurate product claims. The letter mandates correction within a specified timeframe. FDA monitors the company’s corrective actions for adequacy. Subsequent interactions between the FDA and the recipient may impact the issues raised in the letter. Warning Letters are issued for serious violations or insufficient responses to FDA 483s, potentially leading to regulatory action if not resolved effectively.
Example: https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/warning-letters

The FDA Form 483, officially known as “Notice of Inspectional Observations” and commonly referred to as “483,” is issued at the end of an on-site inspection of regulated facilities by the FDA field investigator in response to deficiencies in the quality system or conditions that violate the Food, Drug, and Cosmetic Act.
The 483 does not represent an FDA determination of violations; it is intended to guide corrective actions to be taken by the manufacturer based on inspection. When a manufacturer receives a 483, they must respond FDA by understanding its significance in pharmaceuticals to correct the issue to avoid a warning letter. The FDA 483 form serves as a tool to assist manufacturers in understanding and correcting compliance issues and is the starting point of communication between the FDA and the facility.
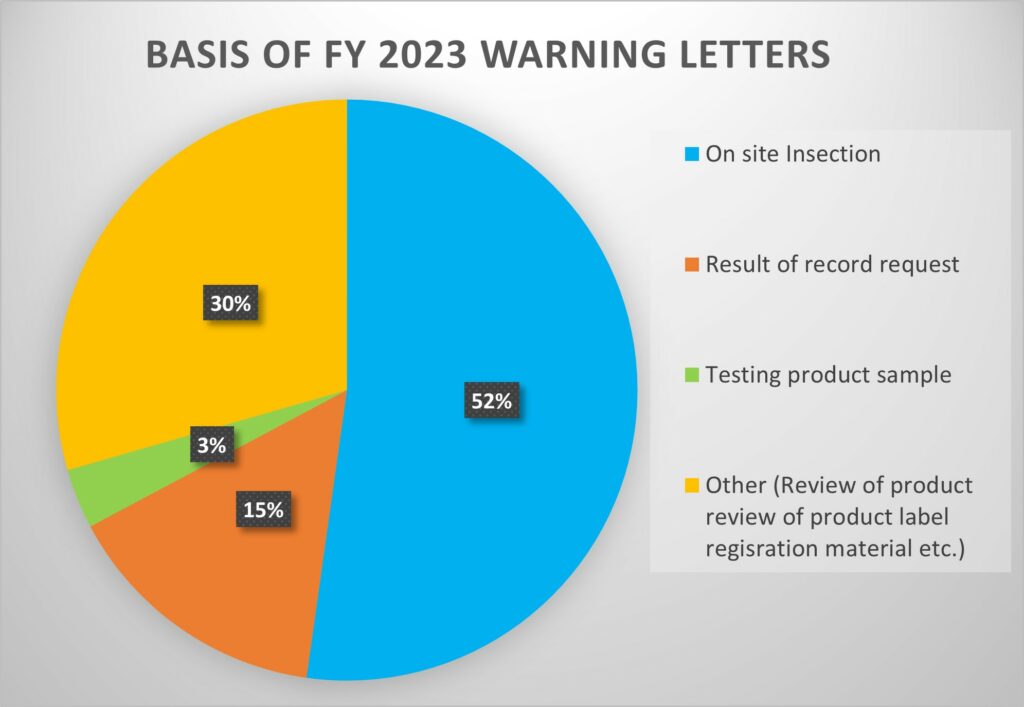
Based on data provided on FDA website, https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/compliance-actions-and-activities/warning-letters
In fiscal year 2023 (FY23), the U.S. FDA issued 180 warning letters to drug and biologics manufacturers. Among these, it happened as below,
- 94 were based on on-site inspections,
- 27 were prompted by records requests,
- 6 were originated from product sample testing
- Others related to product labels, registration materials, and/or websites.
This marked an increase compared to the 165 warning letters issued in FY22, with 74 from inspections.



