
Wie in diesem Blog erläutert, stellt die FDA jedes Jahr Hunderte von Warnschreiben zusammen mit Inspektionen von Produktionsanlagen aus. Sie sollten die Warnschreiben ernst nehmen, denn sie sind ein Vorbote für ein schlimmeres Versagen. Sie sollten stets angemessene Korrektur- und Vorbeugungsmaßnahmen ergreifen und eine umfassende Lückenanalyse auf der Grundlage der genannten Anforderungen durchführen, um die Anforderungen vollständig zu erfüllen.Die Reaktion auf USFDA-Warnschreiben ist wichtig.
FDA-Warnschreiben folgen in der Regel auf Inspektionen, bei denen Abweichungen oder Verstöße gegen Vorschriften, z. B. in Bezug auf die Datenintegrität, festgestellt werden. Während der Inspektionen dokumentieren die Prüfer diese Abweichungen mit dem Formular 483. Die Formulare 483 und die FDA-Warnschreiben sind zwar unterschiedlich, aber das Warnschreiben folgt oft als nächster Schritt im Regulierungsprozess.
Das FDA-Warnschreiben beschreibt den Verstoß und das potenzielle Problem, gefolgt von der Angabe, welche Beweise der Hersteller vorlegen muss, um die angesprochenen Bedenken zu dokumentieren.
Wenn ein Hersteller in erheblichem Maße gegen FDA-Vorschriften verstößt, erhält er ein Warnschreiben der FDA, in dem der Verstoß beschrieben wird, z. B. schlechte Herstellungspraktiken oder ungenaue Produktangaben. In dem Schreiben wird eine Korrektur innerhalb eines bestimmten Zeitrahmens gefordert. Die FDA überwacht die Korrekturmaßnahmen des Unternehmens auf ihre Angemessenheit. Nachfolgende Interaktionen zwischen der FDA und dem Empfänger können sich auf die in dem Schreiben angesprochenen Probleme auswirken. Warnschreiben werden bei schwerwiegenden Verstößen oder unzureichenden Antworten auf FDA 483 ausgestellt und können zu behördlichen Maßnahmen führen, wenn sie nicht wirksam gelöst werden.
Beispiel: https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/warning-letters

Das FDA-Formular 483, offiziell bekannt als „Notice of Inspectional Observations“ und allgemein als „483“ bezeichnet, wird am Ende einer Vor-Ort-Inspektion von regulierten Einrichtungen durch den FDA-Ermittler vor Ort ausgestellt, um auf Mängel im Qualitätssystem oder auf Bedingungen zu reagieren, die gegen das Lebensmittel-, Arzneimittel- und Kosmetikgesetz verstoßen.
Die 483 stellt keine Feststellung der FDA über Verstöße dar, sondern dient als Anleitung für Korrekturmaßnahmen, die der Hersteller aufgrund der Inspektion ergreifen muss. Wenn ein Hersteller ein 483-Formular erhält, muss er darauf reagieren, indem er die Bedeutung des Formulars für Arzneimittel versteht und das Problem behebt, um ein Warnschreiben zu vermeiden. Das FDA-Formular 483 dient als Hilfsmittel für Hersteller, um Probleme bei der Einhaltung der Vorschriften zu verstehen und zu beheben, und ist der Ausgangspunkt für die Kommunikation zwischen der FDA und der Einrichtung.
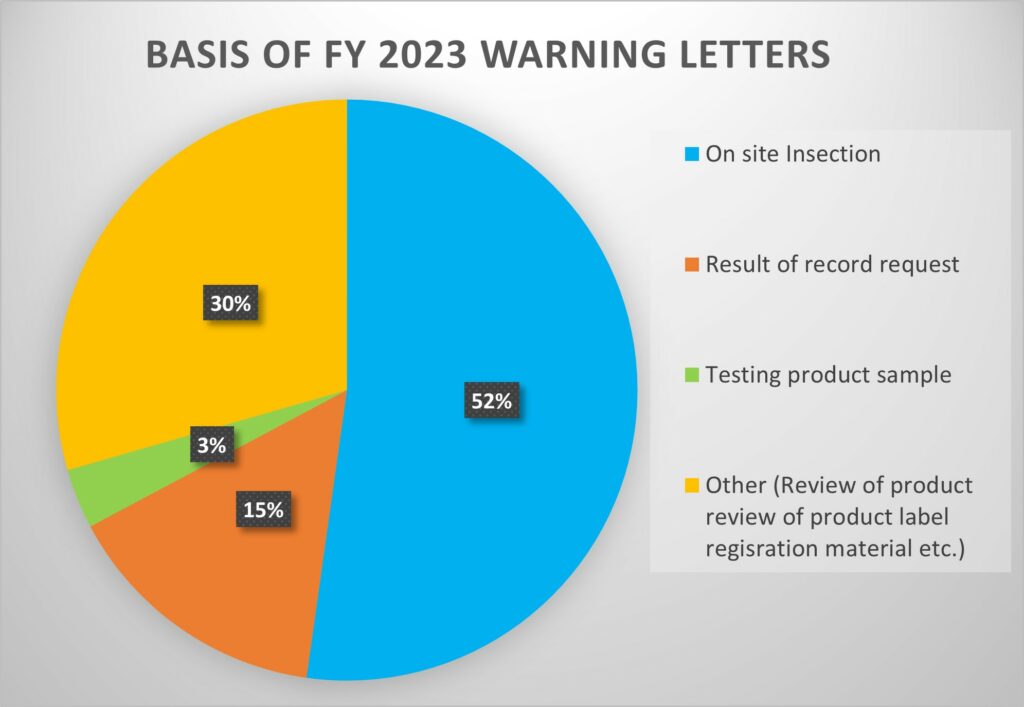
Auf der Grundlage von Daten : : https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/compliance-actions-and-activities/warning-letters
Im Haushaltsjahr 2023 (FY23) hat die US-FDA 180 Warnschreiben an Hersteller von Arzneimitteln und Biologika verschickt. Diese verteilten sich wie folgt,
- 94 basierten auf Inspektionen vor Ort,
- 27 wurden durch Anfragen nach Unterlagen veranlasst,
- 6 stammten aus der Prüfung von Produktproben
- Andere betrafen Produktetiketten, Registrierungsunterlagen und/oder Websites.
Dies bedeutet einen Anstieg im Vergleich zu den 165 Mahnschreiben, die im GJ22 ausgestellt wurden, von denen 74 auf Inspektionen zurückgingen.



